
Qu'est-ce que Sturge Weber ? syndrome?
Le syndrome de Sturge-Weber est rare. congénitalet non hérité vasculaire trouble caractérisé par capillaire malformation sur la peau du visage (tache de vin) et capillaireveineux malformations du cerveau et des yeux [1].
Syndrome de Sturge-Weber
Syndrome de Sturge-Weber
Qui obtient le syndrome de Sturge-Weber?
On estime qu'environ un bébé sur 20 à 50 000 naît avec le syndrome de Sturge-Weber [2]. Comme il est causé par une mosaïque mutationIl n'est pas hérité des parents.
Qu'est-ce qui cause le syndrome de Sturge-Weber?
Le syndrome de Sturge-Weber est causé par une somatique mutation mosaïque de la GNAQ gène dans chromosome 9 9 [3]. Cela signifie que la mutation du gène s'est produite dans les cellules du corps après la formation du zygote. GNAQ réglemente intracellulaire Voies de signalisation. La mutation entraîne la formation ou la maturation incontrôlée de capillaires dans les cellules affectées
Quelles sont les caractéristiques cliniques du syndrome de Sturge-Weber ?
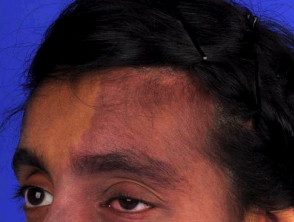
Le syndrome de Sturge-Weber est caractérisé par des malformations vasculaires du visage et du cerveau des personnes atteintes.
- Les taches de vin de Porto sont le type le plus courant de malformation vasculaire, affectant environ trois bébés sur 1 000, mais la plupart ne sont pas associées au syndrome de Sturge-Weber [4].
- Les taches de vin dans le syndrome de Sturge-Weber sont généralement sur le Distribution des première et deuxième divisions du nerf trijumeau dans le front et la paupière supérieure [5]. Ils peuvent également affecter les deux côtés du visage. [6].
- Les malformations vasculaires leptoméningées apparaissent dans le cerveau du même côté que la tache de vin.
- Des malformations leptoméningées peuvent également survenir sans tache de vin [7].
D'autres caractéristiques du syndrome de Sturge-Weber peuvent inclure :
- Convulsions et épilepsie
- Hémiparésie et événements ressemblant à un accident vasculaire cérébral
- problèmes de comportement
- défauts du champ visuel et glaucome
- Déficit en hormone de croissance.
Quelles sont les complications du syndrome de Sturge-Weber ?
Les complications du syndrome de Sturge-Weber dépendent de l'étendue des malformations vasculaires et d'autres caractéristiques cliniques. Ils sont extrêmement variables.
Comment diagnostique-t-on le syndrome de Sturge-Weber ?
Le diagnostic du syndrome de Sturge-Weber repose sur la recherche de taches de vin de Porto et de malformations capillaires veineuses leptoméningées. Un diagnostic basé uniquement sur les lésions leptoméningées dépend de l'évolution des symptômes.
S'il y a neurologique symptômes ou constatations image de résonance magnétique (Résonance magnétique) du cerveau est réalisé avec un contraste de gadolinium pour détecter les malformations capillaires-veineuses leptoméningées.
Quel est le diagnostic différentiel pour le syndrome de Sturge-Weber ?
Les caractéristiques d'autres syndromes de malformation vasculaire sont décrites ci-dessous.
-
Le syndrome de Klippel-Trénaunay présente des malformations capillaires plus étendues, touche les membres et le tronc et est associé à hypertrophie du membre atteint.
- Le syndrome de Parkes-Weber se présente avec une grande malformation capillaire dans un membre et une hypertrophie du membre affecté. Il existe plusieurs shunts artério-veineux à débit rapide.
- Le syndrome de Servelle-Martorell, une maladie angiodysplasique congénitale rare, est associé à progressive hypotrophie des membres.
-
Le syndrome de Protée se présente avec asymétrique et la prolifération disproportionnée des parties du corps. Le syndrome résulte d'une mutation activatrice somatique dans le AKT1 oncogène.
-
Le syndrome de CLOVES se présente avec une prolifération lipomateuse congénitale, une malformation vasculaire, épidermique naevus, anomalies vertébrales/squelettiques/scoliose. Le syndrome résulte de l'activation de la mosaïque somatique. mutations dans le PIK3CA gène.
Quel est le traitement du syndrome de Sturge-Weber ?
Il n'y a pas de traitement spécifique pour le syndrome de Sturge-Weber. Dépend de la gestion cutanéneurologique et oculaire symptômes, avec un succès limité.
Le traitement des crises chez les patients atteints du syndrome de Sturge-Weber avec des antiépileptiques n'est pas toujours efficace [8]. Aucun traitement ne semble être plus haute à d'autres.
Les taches de vin de Porto peuvent être traitées avec un colorant pulsé Être. Les blessures au visage et au cou produisent des résultats pires que les blessures à d'autres parties du corps. [9].
Le glaucome chez les patients atteints du syndrome de Sturge-Weber est traité chirurgicalement et médicalement [10].
Les bébés diagnostiqués avec le syndrome de Sturge-Weber doivent être traités avec de l'aspirine à faible dose [11,12]. Le traitement antithrombotique peut empêcher la progression de la maladie, ce qui peut affecter le flux sanguin vers le cerveau et entraîner des lésions nerveuses.
Quelle est l'issue du syndrome de Sturge-Weber ?
Les prévoir Le syndrome de Sturge-Weber dépend du degré d'atteinte cérébrale et cutanée. Les grandes taches de vin de Porto sont associées à un risque accru d'épilepsie et de glaucome, tandis que bilatéral Les malformations vasculaires leptoméningées sont associées à l'apprentissage et à la déficience intellectuelle. [13]. L'apparition des crises avant l'âge d'un an a un effet significatif sur les fonctions cognitives et motrices chez les enfants atteints du syndrome de Sturge-Weber [14].
On estime qu'environ un adulte sur deux atteint du syndrome de Sturge-Weber présente des anomalies neurologiques, même chez ceux qui ont été initialement asymptomatique.
